BREAKING NEWS
Launching 1st March 2023. Also check out: https://www.thailandmedical.news/
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs) are dreaded neurological diseases with a progressive course, and are invariably fatal.
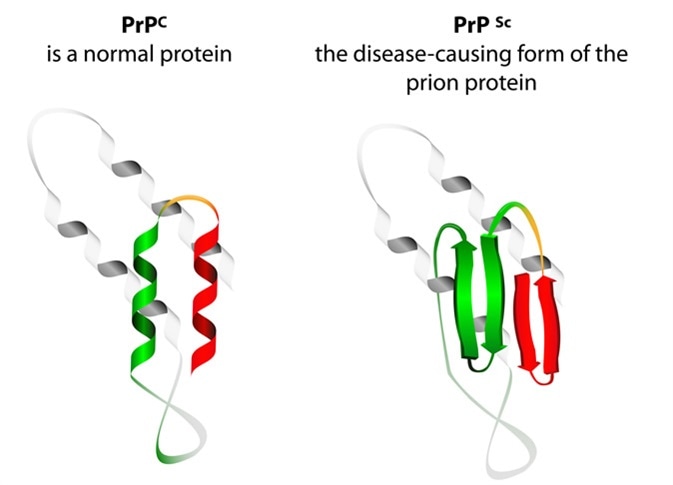
They are caused by tiny particles primarily composed of a pathogenic protein which is actually an abnormal variant of the PrP protein, a normal component of the cell membrane.
Prions are unique in being proteinaceous particles without nucleic acids which show the property of being transmissible.
These proteins are encoded by the host and are identical to the normal PrP protein in their primary structure, i.e. the amino acid sequence. However, following translation, unknown changes occur which result in secondary and tertiary misfolding. The changes are caused by point mutations at certain gene loci on the PrP gene.
TSEs are well known to occur in both humans and animals. The most common human prion disease is Creutzfeldt-Jakob disease (CJD).
Animal TSEs include sheep scrapie, transmissible mink encephalopathy, chronic wasting disease (CWD) of wild ruminants, and bovine spongiform encephalopathy (BSE). These are transmitted within the same species by contact with infected animals or by eating food contaminated with these prions.
Scrapie and CWD spread with ease among the animals in the same population, but BSE and CJD with much greater difficulty.
Some of the factors affecting such transmission include:
BSE prions were known to also spread to other species such as cats, goats, and humans from the original cattle host, by consumption of infected food. In humans, this results in variant CJD. This has forced scientists to recognize that prions need not be specific to a single species.
This has been confirmed by the ease with which BSE spreads to mice though not to hamsters.
Transgenic experiments have shown that transferring genes from one species to another leads to the conferring of susceptibility to clinical prion disease which normally occurred only in the original species from which the gene was taken.
Of interest, human beings with homozygous Codon 129 encoding in the PrP gene are more likely to be vulnerable to sporadic, iatrogenic and variant CJD.
The most common route of infection is direct intracerebral injection. This has occurred largely by experimental inoculation.
Iatrogenic transmission has occurred, from electrodes inadvertently used in patients with unrecognized CJD and later subjected to standard disinfection before use in other patients.
These have been linked to the development of CJD in both patients and animal controls. Cadaveric human growth hormone, as well as dural and corneal grafts, have also been associated with CJD in the recipient.
However, all TSEs are not the same, and ingestion is another common route. BSE, for instance, may more commonly cause infection by ingestion.
Following exposure through peripheral sites, the prion may be detected in lymphoid tissue in the early part of the incubation period, but not in the central nervous system (CNS) until the later part.
Some parts of the body seem to accumulate heavy concentrations of infective prion particles before the first sign of disease occurrence. These include the CNS and the retina.
The sequence of infectivity is thought to be probably as follows:
However, lymphatic dissemination of prions is variable, as it is rarely involved in BSE. This is also influenced by the PRNP genotype of the host.
Prions also spread by blood, from the lymphoid cells which patrol the whole circulation. Researchers have found them to be excreted in urine, though at concentrations 10 times less than that in plasma. It is thought to be increased by nephritis.
Other routes of transmission include:
Thus, oral and parenteral routes are known to cause transmission. The exact mechanism whereby some members who feed upon contaminated material develop the disease is unknown as of now, but may involve digestive tract lesions in addition to the presence of prions themselves.
Once they cross the intestinal epithelial barrier, they multiply in the intestinal lymphoid tissue and the enteric nervous system, as well as other lymphoid sites, especially the spleen.
Mature follicular dendritic cells are preferentially utilized to amplify these particles following which PrPSc accumulation occurs, and this requires mature B cells to be present, but other routes of prion amplification are also present.
Finally, the prions move along the peripheral nerves and reach the brain and spinal cord via the vagus or sympathetic nerves. Direct neuroinvasion may also occur if a very high prion dose reaches the peritoneal cavity, as during surgery with contaminated tools.