BREAKING NEWS
Launching 1st March 2023. Also check out: https://www.thailandmedical.news/
Prion diseases are progressive nervous system diseases which always have a fatal outcome. They are transmissible and affect many mammals. They are also called transmissible spongiform encephalopathies (TSEs).
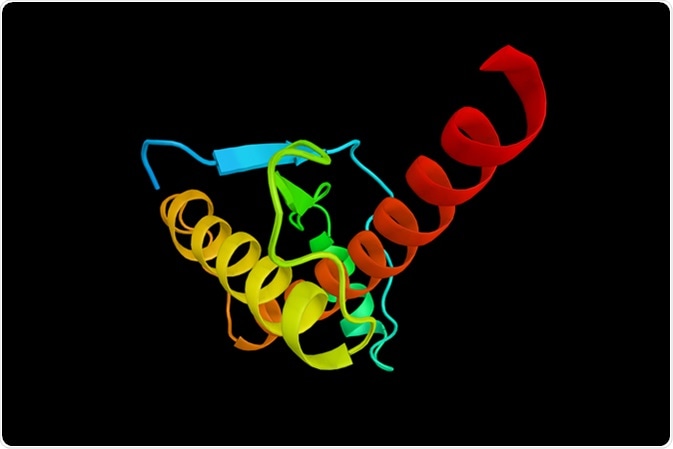
Their root disorder is the abnormal tertiary folding of a protein encoded by the host cells which leads to its aggregation. This is the prion protein, PrP or PrPc, which is transformed into the pathogenic form PrPSc . Both human and animal prion diseases are known: animal TSEs include scrapie, feline spongiform encephalopathy, transmissible mink encephalopathy, chronic wasting disease and bovine spongiform encephalopathy (BSE).
Prion diseases in humans may be sporadic, hereditary or acquired. The sporadic forms include:
Genetic prion diseases, formerly called familial, make up about 10-15% of the total, and are due to autosomal dominant PrPc mutations on the PRNP (PrP encoding gene), and include:
Acquired prion diseases are the least common (5%), and include:
The PrPSc and PrPC forms of the protein are different in secondary and tertiary protein structures but the primary structure is conserved. The abnormal isoform contains beta-helix conformations as opposed to the alpha-helical folding of the normal protein. The result is a protein which resists degradation by most chemical and physical agents used for sterilization, quite unlike the normal protein. It tends to clump with other PrPSc proteins and strongly bind to the normal prion protein. This induces the misfolding of normal PrPc as well, in a rapidly spreading process which leaves the brain dotted with areas of neuronal loss.
This is the most common form of CJD (85%) and causes about 1-2 cases/million. Most cases occur in the individuals between the age of 55 and 75 years. It affects males and females equally. The symptoms include dementia which worsens rapidly, myoclonus, cerebellar symptoms including incoordination of muscular movement, and difficulties with speech, vision and gait. Spastic symptoms indicating pyramidal and extrapyramidal involvement, as well as behavioral changes are common. The final stage is of unresponsiveness called akinetic mutism. The characteristic change is called spongiform encephalopathy, because the abnormal protein clumps within the brain, causing loss of neurons, leaving holes in it like a sponge. This is associated with a low number of PrPSc lesions in the CNS.
This was formerly termed thalamic dementia, and is caused by a mutation of the PRNP gene. It occurs equally in both sexes, and peak incidence is between the age of 20 and 72 years. The average lifespan after the onset of symptoms is 18 months. It begins with insomnia, often accompanied by myoclonus, gait and speech problems, pyramidal symptoms and autonomic hyperactivation. The chief diagnostic instrument is polysomnography.
The condition appears similar to FFI but without a family history or detectable PRNP mutations.
This is caused by point mutations resulting in insertions or deletions of octapeptide repeats in the PRNP gene. However, in more than half of these cases, there is no positive family history.
This autosomal dominant condition is caused by PRNP mutations and occurs between the age of 30 years and 60 years. The symptoms include cerebellar, speech, vision and muscular movement abnormalities, and dementia. The brain shows numerous amyloid lesions, astrocytosis, and neuronal loss. Spongiform changes ranging from nil to abundant are also observed.
The first such case was seen in 1974 following the insertion of a cadaveric corneal graft taken from a person with CJD. Most succeeding cases have occurred due to contaminated stereotactic EEG needles, other neurosurgical instruments, cadaveric dural grafts and cadaveric human growth hormone or gonadotropin derived from the pituitary gland.
This prion disease is almost only seen among the Fore group and closely related tribes, in Papua New Guinea, who used to practice cannibalism as a ritual. Women and children were at higher risk as they ate the brain and viscera compared to the men who ate the muscle tissue by choice. The disease is thought to be due to transmission of sCJD from the dead person by cannibalistic consumption.
At its worst, 1-2 persons in every 100 of the population fell victim to the condition, which in some villages killed all the women. Now it is almost extinct, being confined to some older patients who contracted it before the current ban on the practice but became symptomatic only 5 decades later. Its prevalence has led to a high frequency of Codons 127 and 129 on the PRNP gene in this population because of their association with disease resistance.
It has three stages, the ambulant, sedentary and terminal stages, often with a prodromal stage. The signs are cerebellar ataxia and involuntary movement disorders. A prominent shivering which worsens on exposure to cold is the origin of the name “Kuru”. Dementia is usually a terminal manifestation. Spongiosis and neuronal atrophy with gliosis of the astrocytes are seen in brain tissue. Kuru plaques are noticeable, being spherical bodies from which filaments radiate.
This was first described in 1996 as a form of CJD in which psychiatric symptoms were predominant. The patients were markedly younger, with the incidence being highest between the age of 16 years and 39 years.
This form was first recognized in 2008 as atypical dementia in the patients who were homozygous for PRNP Codon 129 mutations with a family history of cognitive impairment. The brain lesions were characteristic of other prion diseases, but the disease duration was about 24 months longer.